Avogadro Tutorial
This tutorial will guide you through building a peptide in Avogadro and preparing it for use in simulations, with attention to chirality editing for D-amino acids.
Step 1: Open Avogadro and Build Peptide
-
Launch Avogadro.
-
Navigate to the top menu: Build → Insert → Peptides
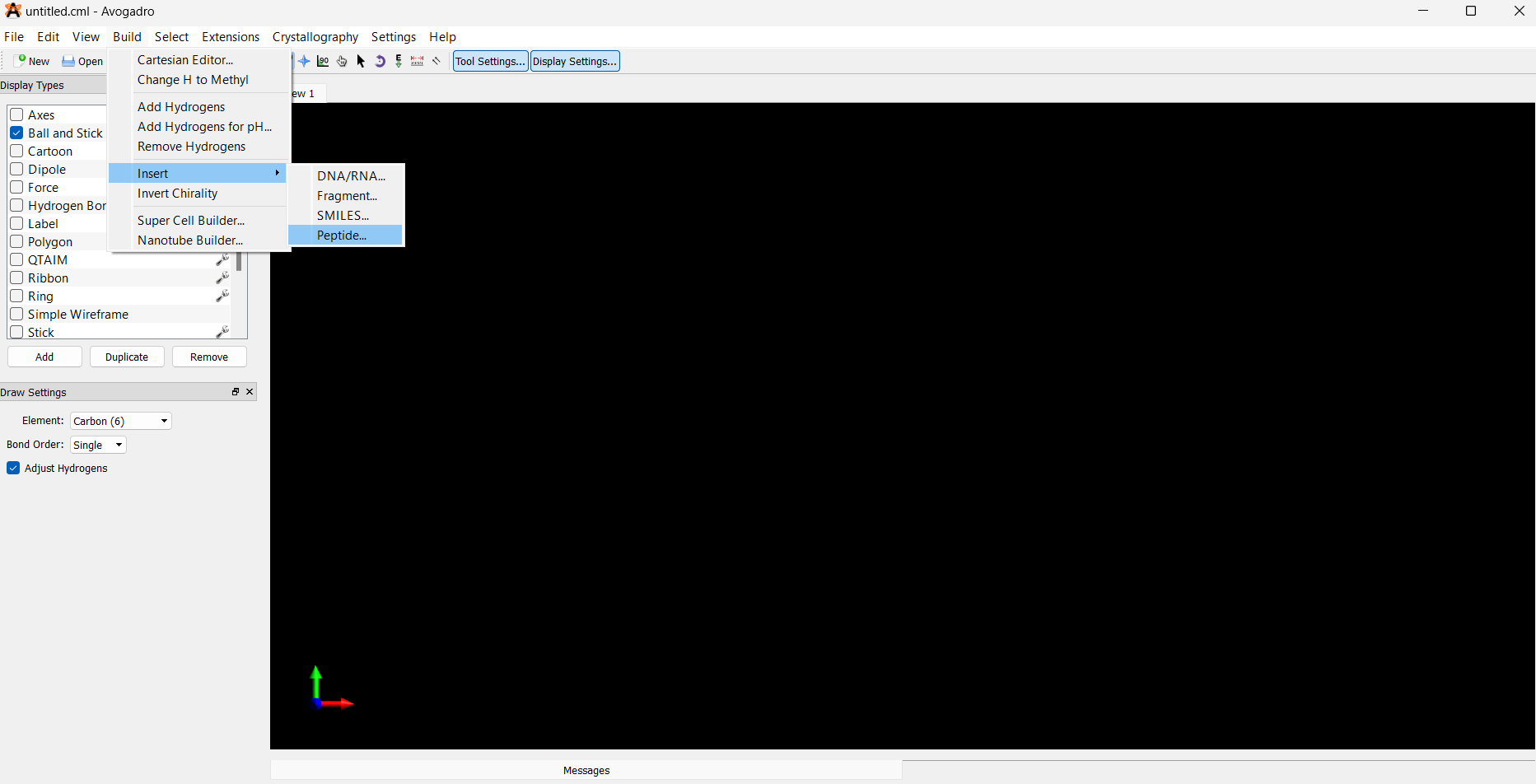
- In the Insert Peptides window, click to add the desired amino acids to construct your peptide.
Example:
KYFIL→ Lys-Tyr-Phe-Ile-Leu
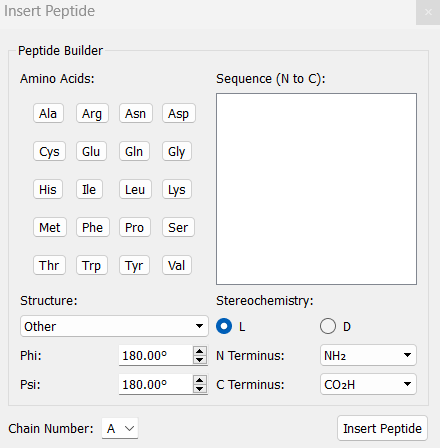
Step 2: Modify to D-Stereochemistry (if needed)
For D-amino acids, you must manually invert chirality:
- Deselect the entire structure: Select → Select None
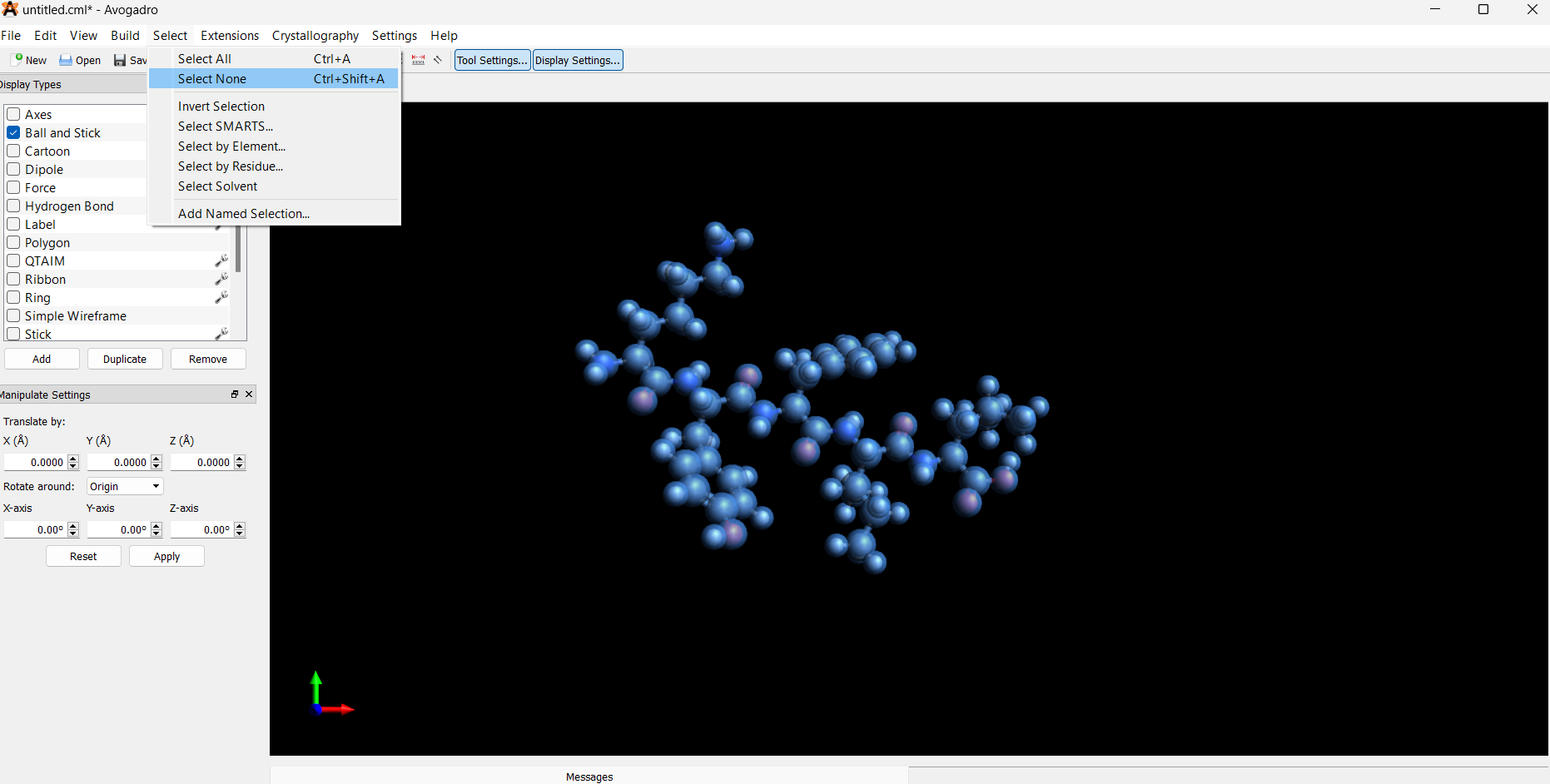
-
Manually select the α-carbon on the amino acid residue (and β-carbon for Ile):
-
Only select the α-carbon and adjacent β-carbon for each D-amino acid.
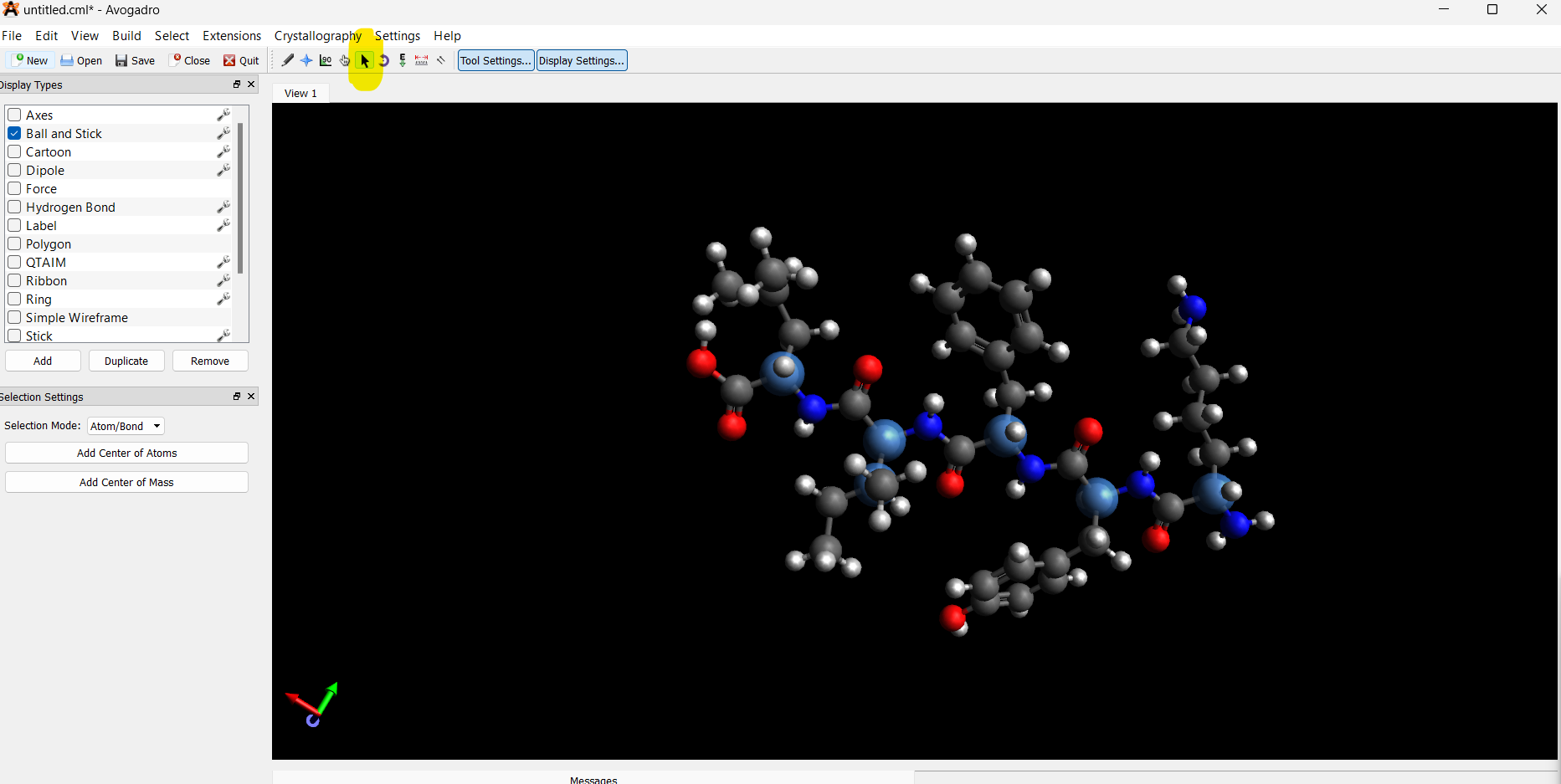
- Invert chirality: Build → Invert Chirality (or right-click if using tools)
Repeat for each D-residue. Avogadro does not automatically label chirality, so this step is crucial for accuracy.
Step 3: Save and Modify PDB File
After saving your .pdb file:
-
For D-residues, manually update the residue names in a text editor:
-
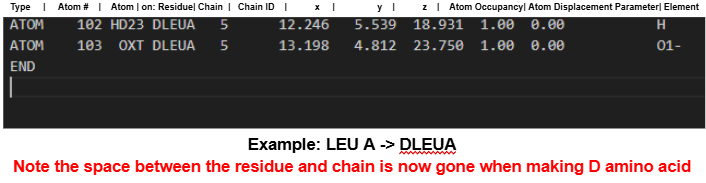
Change 3-letter amino acid codes to include a D prefix (e.g.,
TYR→DTYR). - Ensure there are no spaces in the updated codes.
Step 4: Use CHARMM-GUI for Further Processing
-
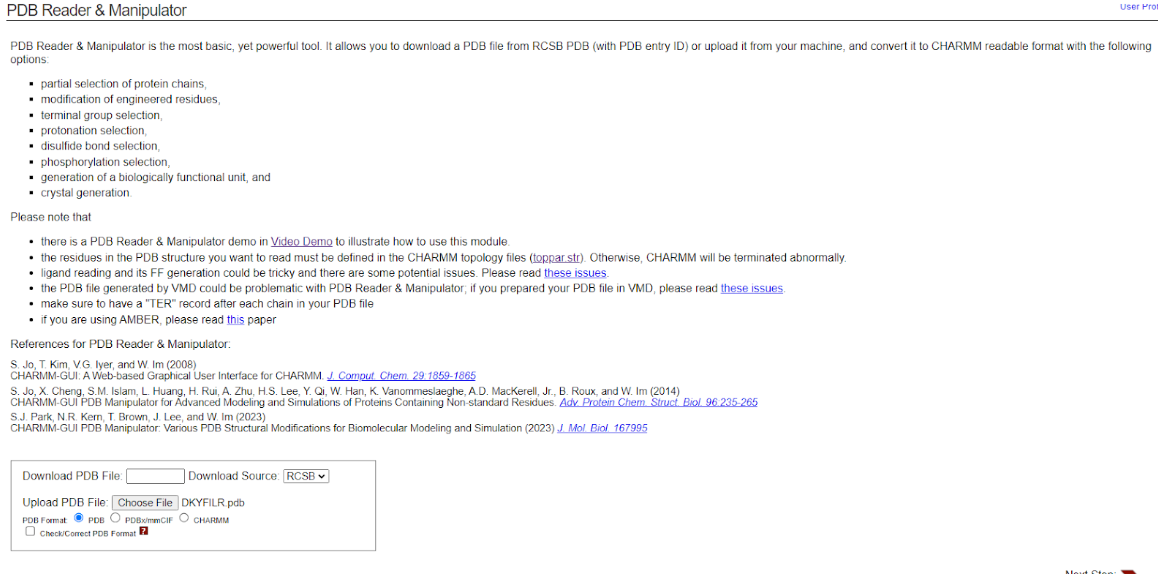
Go to CHARMM-GUI PDB Reader.
-
Upload your edited PDB file under PDB Reader & Manipulator.
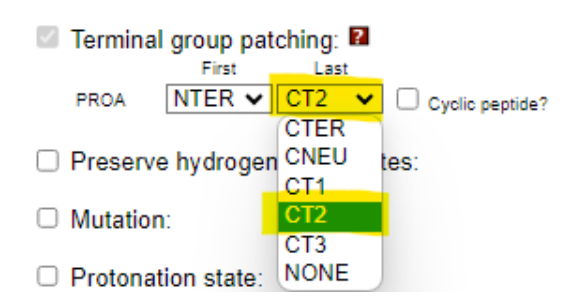
- Leave the default settings unchanged.

- Amidate the C-terminus by checking the appropriate box.
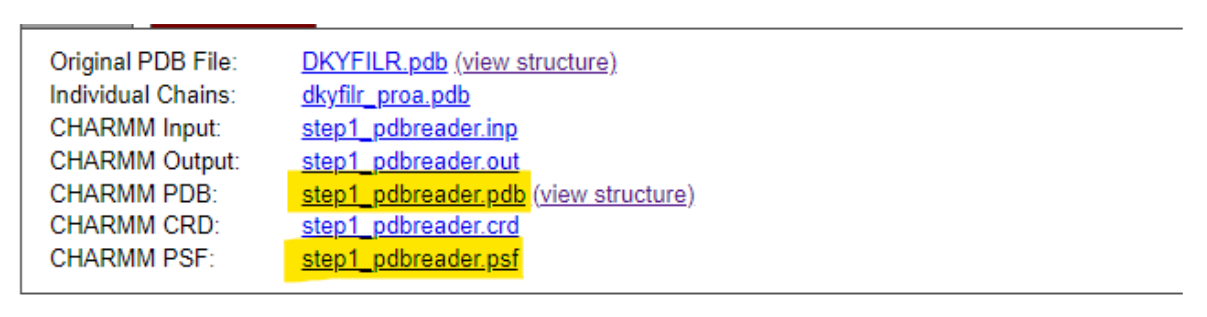
- Download the processed files (e.g., PSF and PDB).
- Open the downloaded
.pdbfile and reapply the D-prefix to any D-residues if CHARMM-GUI reverted them.

Notes
- For L-amino acids, you do not need to invert chirality or change residue codes.
-
After generating your final structure, proceed to the appropriate tutorial:
-
For atomistic simulations, use the NAMD or GROMACS workflow.
- For coarse-grained simulations, continue to the Coarse Grain with GROMACS Tutorial.